Deep within the surface of nearly every human cell lies a vast communication network—tiny molecular sentinels constantly listening, responding, and adjusting the body’s internal balance. Among the most important of these are G-protein-coupled receptors (GPCRs), one of the largest families of cell-surface proteins known. They recognize signals ranging from hormones to neurotransmitters to drugs, translating chemical messages into biological action.
These receptors are not obscure biological curiosities. They regulate many of the processes that sustain everyday life. Even more striking, they serve as targets for over 30% of currently marketed drugs. When medicines influence how we breathe, stay awake, respond to allergens, or control inflammation, GPCRs are often the molecular gatekeepers involved.
Within this immense receptor family sits the histamine H1 receptor (H1R), a protein that quietly orchestrates some of the body’s most familiar experiences. It plays a central role in allergic reactions, inflammation, vascular permeability, airway constriction, wakefulness, and even cognitive functions. Antihistamine drugs primarily target this receptor. Yet despite decades of use, current medications do not always deliver ideal therapeutic outcomes. Their effectiveness can be limited, prompting scientists to ask a deeper question: what if understanding how drugs bind is just as important as knowing that they bind?
A New Way of Seeing Drug Binding
Traditionally, drug design has focused heavily on binding affinity—how tightly a compound attaches to its target protein. But binding is not a single-dimensional event. It is shaped by the delicate balance of enthalpy and entropy, two thermodynamic forces that together determine the energetic landscape of molecular interaction.
Enthalpy reflects the strength of interactions such as chemical bonds and molecular attractions. Entropy reflects disorder and flexibility—the freedom molecules gain or lose when binding occurs. Sometimes, when one increases, the other compensates. This phenomenon, known as enthalpy–entropy compensation, has emerged as a crucial concept for understanding why certain molecules bind more selectively or behave differently despite similar overall binding energies.
Yet directly measuring these thermodynamic components has remained technically challenging, especially for complex cell-surface proteins like GPCRs. This gap has left researchers with an incomplete picture of how many drugs truly interact with their biological targets.
A team led by Mitsunori Shiroishi at Tokyo University of Science in Japan set out to change that. Working alongside researchers including Hiroto Kaneko and Tadashi Ando, the group conducted a systematic investigation into the thermodynamics of H1R binding. Their findings appeared online in ACS Medicinal Chemistry Letters on January 26, 2026.
Their goal was ambitious but precise: to reveal how subtle molecular differences reshape the energetic choreography between a drug and its receptor.
A Familiar Drug, Seen From a New Angle
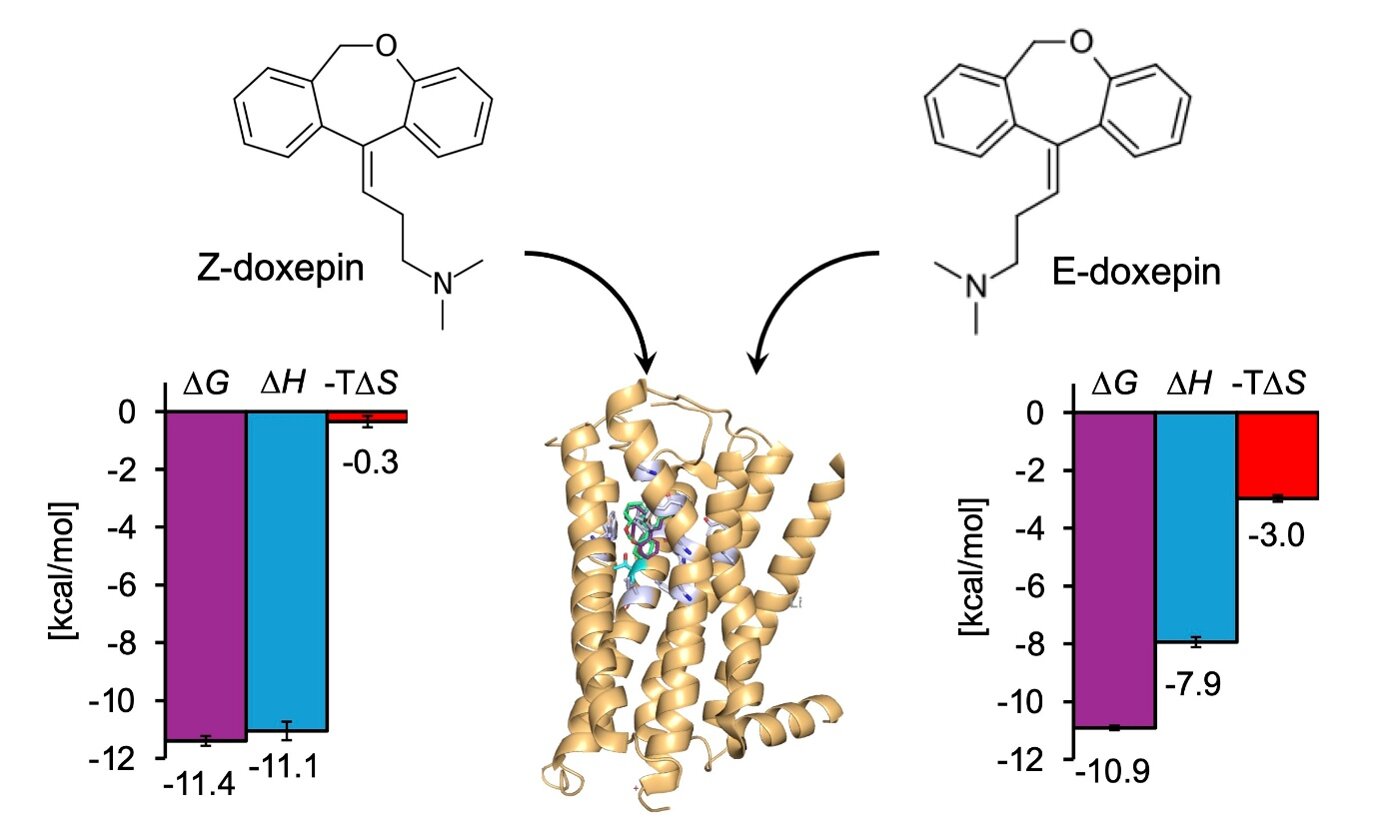
At the center of the investigation was doxepin, a compound widely used as an H1R inhibitor. Although known primarily as a tricyclic antidepressant, it also functions as a powerful antihistamine. But doxepin is not a single rigid structure. It exists as two geometric isomers, called the E-isomer and the Z-isomer, which differ only in spatial arrangement.
Previously, the research team had discovered something intriguing: the Z-isomer binds to H1R with about five times higher affinity than the E-isomer. Even more striking, they identified a specific amino acid in the receptor—Thr1123.37—that appeared to influence this selectivity.
In their new study, the scientists wanted to understand why this difference exists. Not simply which isomer binds better, but what thermodynamic forces shape that preference.
To do this, they created two versions of the receptor. One was the natural wild-type H1R, previously studied. The other was a modified version in which the key amino acid Thr1123.37 was replaced with a different one, forming a mutant receptor called T1123.37V.
They then measured how mixtures and individual forms of the E- and Z-isomers interacted with each receptor. Using isothermal titration calorimetry, they tracked the heat changes during binding, allowing direct measurement of enthalpy and entropy. At the same time, molecular dynamics simulations provided a moving, atomic-level picture of how the molecules behaved.
Together, these approaches revealed not just how tightly the drug bound, but how the binding process unfolded energetically and structurally.
When Energy Tells a Hidden Story
At first glance, the results seemed deceptively simple. The overall binding energy of doxepin interacting with the natural receptor and the mutant receptor showed no difference. If energy alone were considered, one might assume the interactions were essentially the same.
But when researchers examined the components of that energy, a very different story emerged.
Binding to the natural receptor was primarily enthalpy-driven. Strong molecular interactions contributed heavily to the stability of the complex. In contrast, binding to the mutant receptor showed a reduced enthalpic contribution and a larger entropic contribution. The total energy remained similar, but the balance between order and interaction strength had shifted.
The difference became even more pronounced when examining the individual isomers. The Z-isomer binding to the natural receptor produced a larger gain in enthalpy but also a greater loss of entropy compared to the E-isomer. In other words, it formed stronger interactions but at the cost of reduced molecular freedom.
Remarkably, this distinction disappeared in the mutant receptor. There, both isomers behaved similarly, and their binding energies became comparable. The specific amino acid Thr1123.37 had been quietly orchestrating a thermodynamic balancing act—especially for the Z-isomer.
This revealed something profound. A single residue in a receptor can finely tune how much order and interaction strength are exchanged during binding. It does not merely affect whether a drug binds, but how the entire energetic trade-off unfolds.
The Shape of Binding in Motion
Numbers alone cannot capture the full complexity of molecular behavior. To understand why the Z-isomer gained more enthalpy while losing more entropy, the researchers turned to molecular dynamics simulations.
These simulations revealed that the Z-isomer’s stronger binding arose from conformational restrictions—it became more tightly constrained when interacting with the receptor. Such restriction reduces entropy because the molecule has fewer possible arrangements. Yet that same rigidity enhances interaction strength, increasing enthalpy.
In essence, the Z-isomer locks itself into a more ordered, tightly interacting state. That locked position stabilizes the binding but limits flexibility. The energetic cost of reduced disorder is compensated by the energetic gain of stronger molecular interactions.
This dynamic explains the observed thermodynamic pattern: high enthalpy paired with reduced entropy. It is a physical manifestation of the enthalpy–entropy trade-off occurring at atomic scale.
The mutation removing Thr1123.37 relaxed this constraint, allowing the isomers to behave more similarly. The delicate balance between flexibility and interaction strength had been altered by a single molecular change.
A New Lens for Understanding Drug Selectivity
The study demonstrates that tiny structural differences—whether in a drug molecule or a receptor—can shift the balance between enthalpy and entropy without changing total binding energy. Two interactions may appear equally strong overall while being governed by entirely different physical mechanisms.
This insight reshapes how scientists think about ligand selectivity. Instead of focusing only on binding strength, researchers must consider how molecular flexibility, structural constraint, and energetic compensation shape the final interaction.
By combining thermodynamic analysis with molecular dynamics simulations, the research offers a powerful framework for studying not only H1R but potentially many other proteins. The approach provides a window into the hidden forces that determine why some drugs are more selective, more effective, or longer lasting than others.
Why This Research Matters
This work reveals that drug action is not simply about whether a molecule fits a receptor like a key in a lock. It is about how that fit reshapes molecular motion, redistributes energy, and balances order against flexibility. The study shows that even tiny conformational differences can dramatically influence the entropy–enthalpy balance, guiding selectivity and effectiveness.
Understanding this balance could help scientists design therapeutics with improved selectivity, meaning they interact more precisely with their intended targets. That precision could reduce off-target effects, minimize side effects, and potentially produce longer-lasting treatments. By revealing the energetic logic behind molecular binding, researchers gain the ability to shape interactions intentionally rather than relying on trial and error.
The implications extend beyond a single receptor. The same thermodynamic principles may govern interactions across many proteins and drugs. By mapping how energy and structure intertwine, scientists move closer to rational drug design—a future where therapies are engineered with atomic-level understanding of how they will behave in living systems.
In the quiet interplay between enthalpy and entropy, the study uncovers a deeper truth about biology: the smallest molecular adjustments can ripple outward, shaping how medicines work and how the body responds. What once seemed like invisible thermodynamic bookkeeping may, in fact, be one of the most powerful tools for designing the medicines of tomorrow.
Study Details
Hiroto Kaneko et al, Enthalpy–Entropy Trade-Off Underlies Geometric Isomer Selectivity in Histamine H1Receptor–Doxepin Interaction, ACS Medicinal Chemistry Letters (2026). DOI: 10.1021/acsmedchemlett.5c00696