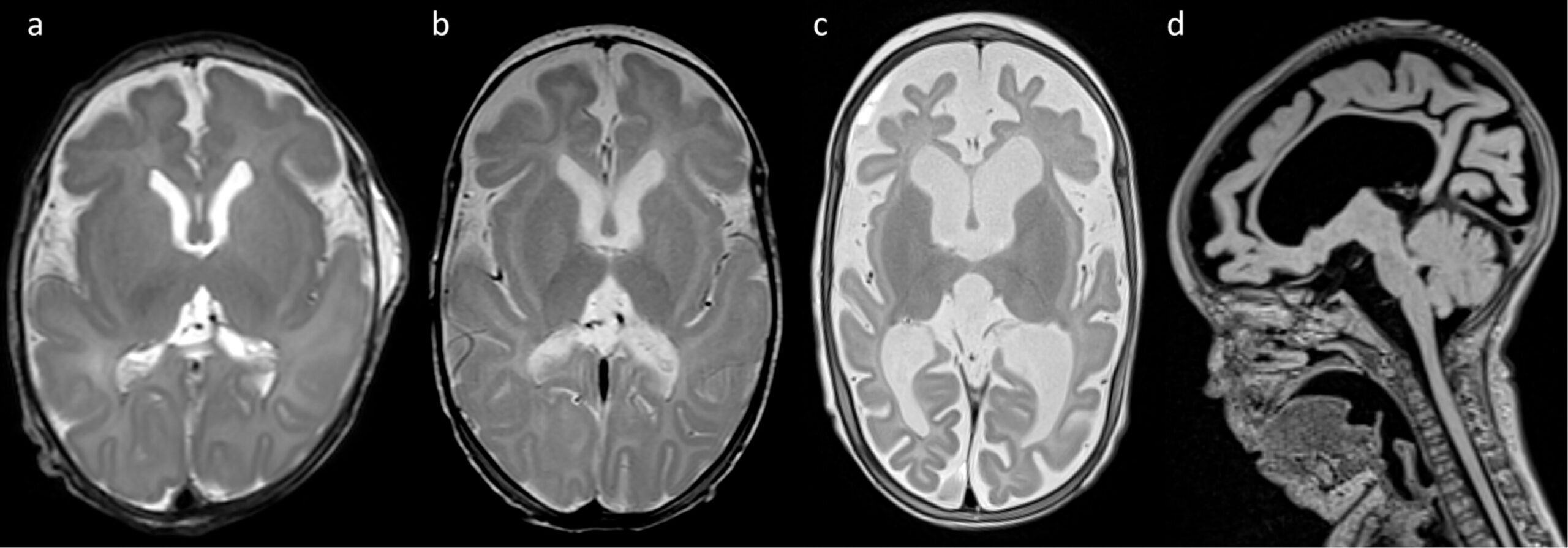
In the hushed chambers of neonatal intensive care, where fragile beginnings unfold under the glow of monitors and machines, a pioneering medical intervention has carved a path through uncertainty. Researchers from Ludwig-Maximilians-Universität München have achieved what was once a distant hope: dramatically reducing seizures in a preterm infant suffering from one of the most severe and intractable forms of epilepsy—SCN2A developmental and epileptic encephalopathy (DEE).
This was not a typical medical trial. There were no groups of patients or placebo arms. There was just one infant, a newborn girl born prematurely at 29 weeks and four days gestation, whose life had been gripped by relentless seizures from the moment she took her first breath. Within hours of birth, she was convulsing up to 25 times an hour. But against daunting odds, her condition was brought under control through a daring and precise molecular strategy using an experimental therapy called elsunersen—a specially designed antisense oligonucleotide (ASO) aimed directly at the root of her disorder.
In a study recently published in Nature Medicine, this single-patient intervention signals not just a beacon of hope for families wrestling with devastating early-onset epilepsies, but a bold stride into the future of precision neuromedicine.
The Genetic Code of Crisis: Understanding SCN2A
To comprehend the breakthrough, we must start with the gene that lay at the heart of this child’s suffering: SCN2A. This gene encodes the Nav1.2 sodium channel, a molecular gatekeeper responsible for the electrical impulses that travel through neurons in the brain. In healthy development, Nav1.2 helps fire action potentials—tiny pulses that transmit information between nerve cells.
But when mutations occur in SCN2A, the consequences can be catastrophic. The gene is highly dosage-sensitive, meaning that both overactivity and underactivity can cause serious disease. Loss-of-function mutations in SCN2A tend to result in later-onset epilepsy or autism spectrum disorders without seizures. In contrast, gain-of-function mutations, where the channel becomes hyperactive, lead to much more severe conditions, often manifesting as DEE within weeks—or even days—of birth.
In these early-onset cases, infants endure dozens of seizures per hour, often resistant to every conventional drug in the medical arsenal. Beyond the seizures lie broader consequences: disrupted brain development, loss of motor control, and cognitive decline. It is a grim and exhausting path for both patients and their families.
A Molecular Scalpel: How Antisense Oligonucleotides Work
For decades, drug development for such genetic epilepsies revolved around symptom management—attempting to control seizures with a growing list of anticonvulsants. But these approaches often did little to improve neurological development, because they did not address the genetic root of the disease.
Enter antisense oligonucleotides, or ASOs. These small, synthetic strands of nucleic acids are built to pair with RNA—the molecular messenger between DNA and protein. By binding to a specific RNA sequence, ASOs can block translation, alter splicing, or trigger degradation of the message. In the case of gain-of-function SCN2A variants, the goal is simple but delicate: reduce the expression of the overactive sodium channel without silencing it completely, thus calming the brain’s electrical storm.
This is the approach taken by elsunersen, a chemically modified “gapmer” ASO designed to selectively bind SCN2A mRNA and trigger its degradation. In preclinical studies using cell cultures and animal models, elsunersen had already shown its ability to lower SCN2A expression and reduce seizure activity. But the leap from bench to bedside is never simple—especially when the “bedside” belongs to a preterm newborn teetering between life and death.
One Child, One Treatment, One Breakthrough
The patient in the study—a premature infant girl born nearly 11 weeks early—was diagnosed with a pathogenic gain-of-function SCN2A mutation based on genetic testing. From the first day of life, she exhibited near-continuous seizure activity, unresponsive to a total of nine conventional antiseizure medications, including heavy-hitters like phenobarbital, phenytoin, levetiracetam, and midazolam. Her condition escalated into status epilepticus—a state of repeated seizures with no recovery in between, often life-threatening.
With conventional medicine at its limits, the research team initiated an emergency therapeutic protocol. At 7 weeks of age, she received her first dose of elsunersen, administered intrathecally—via lumbar puncture—directly into the cerebrospinal fluid that bathes the brain and spinal cord.
The results were nothing short of astounding.
Reversing the Storm: Tracking the Therapeutic Impact
Just eight days after the initial dose, the medical team observed the first significant breakthrough: intermittent breaks in the girl’s otherwise continuous seizures. Status epilepticus began to subside. With each successive dose—meticulously calibrated and spaced based on pharmacokinetic modeling—the seizure frequency dropped further.
Over a 12-week treatment window, three escalating doses totaling 2.5 mg of elsunersen were administered. Seizure frequency fell by 67%, from nearly 20 seizures per hour to just 6.6. With seven doses administered during the acute treatment phase and monthly maintenance dosing afterward (ten additional doses of 8 mg), seizure control stabilized. At 22 months of age, she continued to experience five to seven seizures per hour—still a serious condition, but far from the neurological inferno that had defined her first weeks of life.
Notably, this improvement occurred without any serious side effects. The infant’s vital signs remained stable, and her blood, cardiac, and cerebrospinal fluid profiles showed no abnormalities attributable to the treatment. There were no signs of hydrocephalus, inflammation, or other complications often feared with invasive central nervous system therapies.
Precision in the Face of Fragility: Navigating the Risks
What makes this achievement all the more remarkable is the balancing act required. SCN2A’s role in the brain is critical—not only for excitatory signaling but for maintaining the intricate timing and patterns of neural communication. Suppress too much, and the brain might plunge into lethargy or paralysis. Suppress too little, and seizures continue unabated.
To guide their approach, the research team leveraged advanced tools including voltage-clamp electrophysiology, dynamic action potential protocols, and in silico structural modeling. These methods confirmed the specific dysfunction in the patient’s mutated Nav1.2 channel: increased persistent sodium current and impaired inactivation, consistent with a gain-of-function effect. Based on this understanding, the team was able to model distribution, half-life, and efficacy of the ASO therapy in brain tissue.
They also uncovered an intriguing pharmacodynamic insight: seizure severity tended to increase four to six weeks after each dose, suggesting a gradual waning of the drug’s effect and helping define the appropriate dosing interval. This is a vital discovery not just for this case, but for any future clinical applications of SCN2A-targeted ASOs.
Beyond the Seizures: The Larger Promise of ASO Therapies
While the results of this single-patient study must be interpreted cautiously, they nevertheless mark a significant proof of principle: a rationally designed ASO, targeting a specific genetic mutation, can dramatically reduce seizure burden in early-onset DEE.
But the implications run deeper.
This study reflects a paradigm shift in how we approach neurological diseases—moving from blunt, broad-spectrum medications to personalized, molecularly targeted therapies. Antisense oligonucleotides are now being explored in a range of conditions, from spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS) to Huntington’s disease and Rett syndrome.
In each case, the dream is the same: to intervene at the level of RNA, halting or reversing disease progression at its source rather than merely managing symptoms. For families devastated by early-onset genetic disorders, this approach offers hope that was unimaginable even a decade ago.
One Small Patient, One Giant Leap
The infant girl at the center of this breakthrough is still living with epilepsy, and her future remains uncertain. But thanks to the intervention, she now has a fighting chance—a future measured in smiles and milestones rather than seizures and setbacks.
For her family, the therapy is nothing short of miraculous. For the medical community, it’s a powerful demonstration of what’s possible when genomic insights, chemical precision, and clinical bravery converge. And for the next generation of patients born into similar genetic battles, it could represent a new standard of care.
As formal clinical trials loom on the horizon, researchers continue to refine dosing strategies, monitor long-term safety, and explore combination therapies—particularly with sodium channel blockers that may further fine-tune neuronal excitability.
What began as an emergency rescue mission may ultimately reshape the landscape of pediatric neurology. In the coded language of molecules and genes, a new conversation has begun—one in which silence is not a symptom, but a sign of healing.
Reference: Matias Wagner et al, Antisense oligonucleotide treatment in a preterm infant with early-onset SCN2A developmental and epileptic encephalopathy, Nature Medicine (2025). DOI: 10.1038/s41591-025-03656-0