Every cell in the human body contains the same master script: the DNA that defines our identity, governs our biology, and charts our inherited path through life. For most, this genetic code unfolds silently, giving rise to traits like eye color or blood type. But in some, hidden in the dense script of their genes, lies a time-delayed storm—Huntington’s disease. This rare, devastating condition lurks beneath the surface of family trees, passed quietly from one generation to the next, until the moment it awakens, changing everything.
Huntington’s disease, often abbreviated as HD, is not like catching a cold or developing high blood pressure. It is not born from lifestyle, diet, or even bad luck. It is a genetic certainty for those who inherit the defective gene, a disorder with a chilling level of inevitability. It doesn’t strike in childhood, nor does it show its hand early. Instead, it lies in wait, often until midlife, when symptoms slowly begin to unravel the tapestry of the mind and body.
This disease’s roots lie deep in molecular biology. To understand it is to walk into the very heart of human genetics, where a single mutation can echo across decades, destroying neurons, memories, movement, and identity itself.
The Discovery of a Hidden Killer
Though the symptoms of HD have been described in medical literature for centuries, it wasn’t until the 19th century that the disease received its name. In 1872, American physician George Huntington wrote a detailed account of a peculiar inherited disorder that caused severe involuntary movements, mental decline, and psychiatric disturbances. He noted its distinctive hereditary pattern, how it passed from parent to child, striking families generation after generation. His observations were so precise that the disease was eventually named after him—Huntington’s chorea, a term that would later evolve into Huntington’s disease.
Even then, little was understood about what actually caused the condition. It would take another century and the advent of molecular genetics to identify the true culprit: a mutation in a single gene. In 1993, after decades of research and collaboration among scientists worldwide, the precise gene responsible for Huntington’s disease was discovered on chromosome 4. The gene was named HTT, short for “huntingtin,” and within its sequence lay the secret to one of the most enigmatic and cruel conditions known to medicine.
The Gene with a Repeating Curse
The HTT gene encodes a protein known as huntingtin. In healthy individuals, this protein plays various roles in nerve cells, including cellular transport, signaling, and structural support. But in people with HD, a strange repetition occurs. A segment of DNA within the HTT gene—specifically the trinucleotide sequence CAG—is repeated far more times than it should be.
In normal circumstances, the CAG triplet, which codes for the amino acid glutamine, repeats 10 to 35 times. In individuals with Huntington’s disease, this sequence can be repeated 36 times or more, often extending into the 40s, 50s, or beyond. This excessive repetition causes the resulting huntingtin protein to become elongated and misshapen. Instead of performing its usual functions, it becomes toxic, accumulating in neurons and interfering with their ability to function and survive.
The longer the CAG repeat, the more aggressive the disease. Those with very high repeat counts may show symptoms in childhood or adolescence—a variant known as juvenile Huntington’s disease—while those with fewer repeats may not experience signs until their 50s or 60s. But once symptoms begin, the progression is relentless.
A Battle Inside the Brain
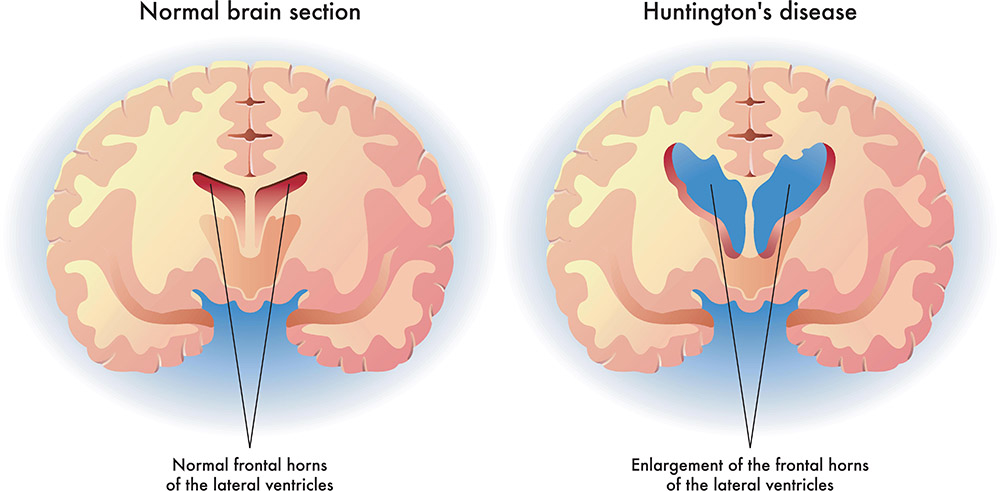
The most tragic aspect of Huntington’s disease is its ruthless attack on the brain, especially the basal ganglia and cerebral cortex. These regions are critical for controlling movement, emotions, and cognition. As neurons in these areas die, the characteristic symptoms of HD begin to emerge.
One of the most visible signs is chorea—a Greek word meaning “dance.” Patients experience sudden, involuntary movements that seem almost playful at first but grow increasingly severe and debilitating. These movements are often accompanied by muscle rigidity, abnormal posture, and difficulty swallowing or speaking.
But HD is not only a movement disorder. It is a disease of the mind. As the damage in the brain spreads, patients suffer from mood swings, depression, anxiety, irritability, and, in some cases, psychosis. Cognitive decline follows closely behind. Memory falters, planning becomes difficult, decision-making erodes, and eventually, the person may lose their ability to recognize loved ones or perform the simplest tasks.
The slow degeneration over 10 to 25 years strips away layers of self, leaving families to witness the heartbreaking transformation of someone they once knew. It is a disease that not only attacks the body but shatters the narrative of a life.
The Cruelty of Inheritance
Huntington’s disease is inherited in an autosomal dominant manner. This means that a person only needs to inherit one copy of the mutated gene—from either parent—to eventually develop the disease. If one parent has HD, each child has a 50% chance of inheriting the mutation. This brutal coin toss has weighed heavily on countless families over generations.
Knowing that one might carry the gene, but not knowing when or if symptoms will appear, creates an unbearable psychological burden. Some individuals choose genetic testing to find out their status, while others prefer to live without knowing. The decision is deeply personal, and the implications are profound. A positive test result is not merely a diagnosis—it is a glimpse into the future, a shadow cast across one’s dreams, career plans, and even decisions about having children.
In families affected by HD, the disease becomes more than just a medical condition—it becomes a presence in the household, an unwanted guest that shapes relationships, choices, and emotional well-being.
Inside the Molecular Machinery
To truly grasp the nature of Huntington’s disease, we must journey deeper into the cellular world, where proteins are built, folded, and managed with extraordinary precision. Proteins are not just strings of amino acids; they are complex, three-dimensional structures that must fold into specific shapes to perform their functions. The huntingtin protein, when distorted by excess glutamine residues from the CAG repeats, misfolds and aggregates into clumps.
These protein aggregates accumulate in neurons, forming inclusions that disrupt cellular processes. They impair communication between brain cells, interfere with the transport of nutrients and organelles, and compromise the health of mitochondria, the energy factories of the cell. The stress on neurons leads to cell death, starting in the basal ganglia and eventually spreading to the cortex and other regions.
Scientists are still unraveling exactly how the toxic huntingtin protein causes this cascade of destruction. Some theories suggest that it disrupts gene transcription and protein regulation. Others point to inflammation and oxidative stress as contributing factors. What is clear is that HD represents a failure of the brain’s normal maintenance and repair systems, overwhelmed by the persistent onslaught of the mutant protein.
Animal Models and Experimental Windows
Because Huntington’s disease progresses slowly in humans and involves complex brain systems, scientists rely heavily on animal models to study it. Genetically modified mice, engineered to carry the human HTT mutation, have been indispensable in advancing our understanding. These mice develop symptoms similar to human HD, including motor dysfunction, cognitive decline, and brain pathology.
Animal models allow researchers to test potential therapies, explore the role of different brain regions, and identify biomarkers that may predict disease onset or progression. Larger animals, including pigs and primates, have also been engineered with the HTT mutation, offering insights into how the disease might function in more complex nervous systems.
These models are not perfect, but they are essential. Without them, the slow march toward a treatment or cure would be even slower. Every experiment, every failed drug trial, and every small breakthrough adds another piece to the puzzle of HD.
From Genetic Clues to Therapeutic Hope
For many years after the gene was discovered, treatment options remained limited to managing symptoms. Physicians could prescribe antipsychotics, antidepressants, or medications to control movement, but these were temporary reprieves. They did not slow the disease, let alone reverse it.
But the tide began to turn with the rise of genetic medicine. Scientists began exploring how to target the mutant huntingtin gene itself. Could the faulty gene be silenced, edited, or replaced?
One of the most promising avenues has been gene silencing therapy. These approaches use molecules like antisense oligonucleotides (ASOs) or RNA interference (RNAi) to prevent the mutant gene from producing its toxic protein. Essentially, they tell the cellular machinery to ignore or destroy the faulty genetic message before it can do harm.
Clinical trials began cautiously. In 2017, the world watched with cautious optimism as Ionis Pharmaceuticals and Roche announced the results of an early-stage trial for an ASO drug known as tominersen. It was one of the first treatments shown to reduce huntingtin protein levels in the brain. Later trials brought mixed results, but they also offered critical information that continues to shape new generations of therapies.
Beyond gene silencing, researchers are exploring CRISPR gene editing, stem cell therapies, neuroprotective compounds, and even small molecules that enhance cellular repair mechanisms. Each of these approaches brings hope, but also caution. The complexity of the brain and the delicate timing of intervention make HD an incredibly difficult target.
Living With the Diagnosis
For those who carry the gene or have been diagnosed with Huntington’s disease, life becomes a balancing act of adaptation, support, and resilience. Patients often need a multidisciplinary team of neurologists, psychiatrists, physical therapists, and social workers. Families, too, become part of the care team, navigating an ever-changing landscape of needs.
Speech therapy may help delay communication difficulties. Occupational therapy can assist with daily living. Counseling and psychiatric care become essential for managing the emotional toll. Support groups and HD advocacy organizations provide a vital lifeline of understanding and community.
In recent years, genetic counseling has become increasingly important. Families faced with the decision to undergo predictive testing need guidance, emotional support, and time to reflect. The choice to know or not know one’s genetic fate is one of the most difficult decisions a person can face.
The Road Ahead: Science with Soul
Huntington’s disease, more than many other conditions, represents the intersection of cutting-edge science and the deepest human emotions. It is a disease that invites awe at the complexity of genetics while demanding empathy for those it affects. Each breakthrough in the lab is shadowed by the real stories of individuals and families living through the experience.
As our understanding deepens, so too does our commitment. Researchers around the world continue to hunt for the key that will unlock a cure—not just for Huntington’s disease, but for the many other neurodegenerative disorders that share similar roots.
What we learn from HD could reshape how we think about the brain, about inheritance, and about our own biological narratives. In the end, the science behind Huntington’s disease is not just about DNA, neurons, or proteins. It is about people. It is about lives suspended between hope and fear. It is about a future that, thanks to science, may yet be rewritten.