For families affected by Tay-Sachs and Sandhoff disease, two rare and fatal childhood disorders, hope has often felt like a distant dream. These conditions, part of a group called GM2 gangliosidosis, rob children of movement, speech and eventually life, often within just a few years. But a clinical trial led by researchers at UMass Chan Medical School is beginning to change that narrative, showing that carefully designed gene therapy can alter the course of these devastating diseases — even if only in early steps.
“This was the first time we could show, biochemically, that the therapy worked,” said Heather Gray-Edwards, DVM, Ph.D., assistant professor of genetic and cellular medicine at UMass Chan and study investigator. “We were able to induce production of the enzyme that these children lack, and that enzyme was functional. Although the levels weren’t yet therapeutic, our thalamic injections were safe and the vectors worked. That is an important step.”
The findings, published in Nature Medicine, mark one of the first demonstrations that gene therapy can produce measurable, functional biochemical correction in patients with Tay-Sachs and Sandhoff disease.
What These Children Face
GM2 gangliosidosis is caused by mutations in genes that normally instruct cells to make beta-hexosaminidase A (HexA), an enzyme responsible for breaking down fatty molecules in the brain. Without HexA, these molecules accumulate inside neurons like toxic waste, gradually destroying them.
Children with the infantile forms of Tay-Sachs or Sandhoff disease typically appear healthy at birth. But within months, subtle delays begin to surface: slower development, weak muscles, and loss of skills they had begun to master. Seizures follow, swallowing becomes impossible, and tragically, most children with infantile forms do not live past early childhood.
There are currently no treatments. For parents, the diagnosis is devastating — a sentence of watching their child fade while having no medical options to intervene. That is why the UMass Chan team’s results are being greeted with such cautious but heartfelt optimism.
How the Therapy Works
The therapy being tested uses a hybrid approach with two harmless viral vectors, carefully engineered carriers that deliver DNA instructions directly into brain cells. These instructions tell the cells how to produce the missing HexA enzyme.
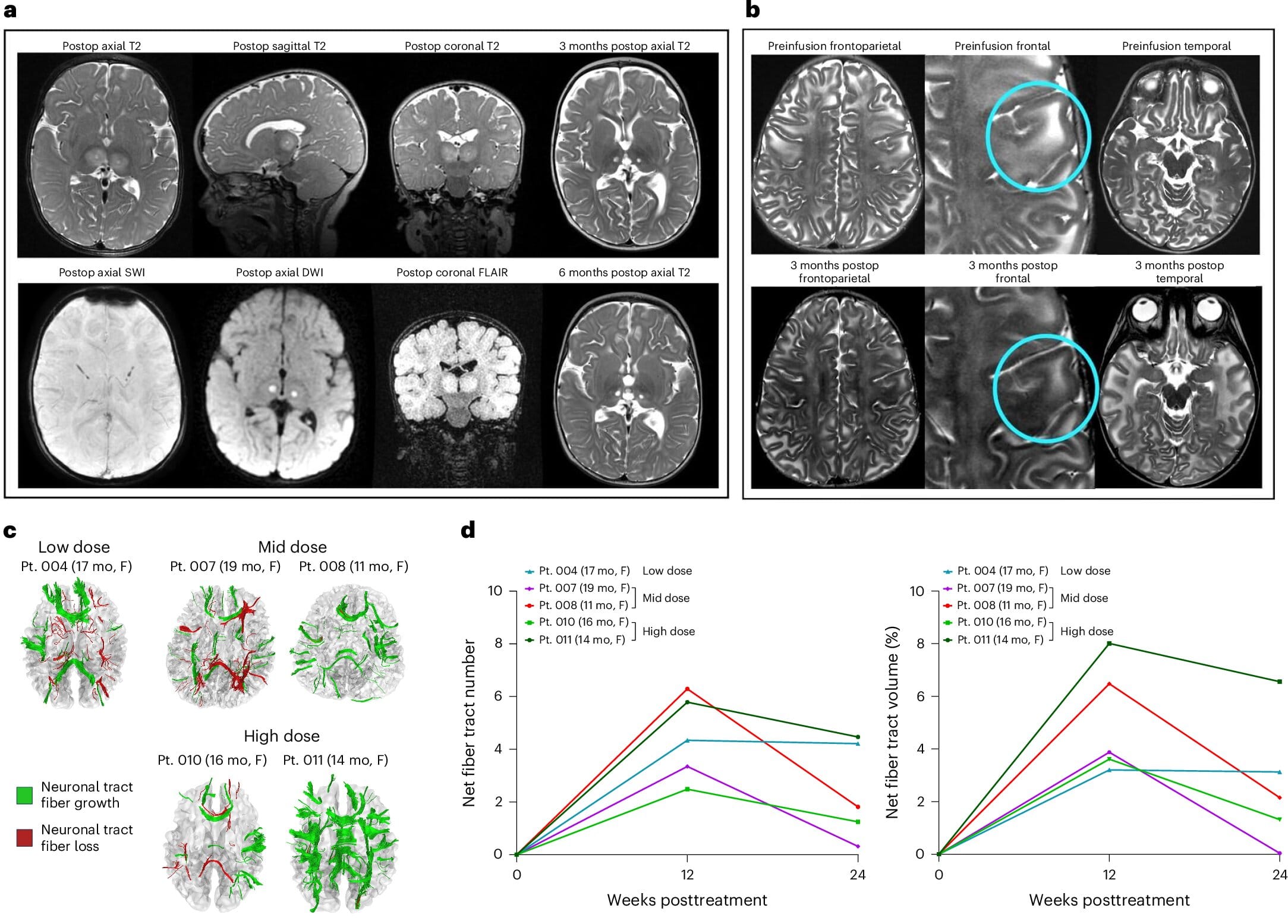
The trial used injections into the thalamus — a central brain region that relays motor and sensory information — as well as into the spinal cord. Once inside neurons, the vectors remain, allowing those cells to continuously manufacture the enzyme. The goal is not a temporary fix but a lasting source of HexA that could halt or slow the relentless buildup of toxic molecules.
Nine participants, divided into four cohorts, received increasing doses of the therapy. Safety was the primary goal of this early trial, but researchers also looked for biochemical and clinical signs of improvement.
Signs of Real, Measurable Change
The outcomes, though preliminary, were meaningful. Laboratory tests showed that all participants began producing HexA, with activity rising to more than twice the lower limit of normal in some cases. For families, however, the most visible improvements came in everyday milestones that matter deeply.
Children in the study who would have normally required feeding tubes before their second birthdays were able to continue eating by mouth. Historically, more than half of GM2 gangliosidosis patients require intravenous feeding between 13 and 18 months. In this trial, half of the children maintained full oral feeding for at least 25 months, and the two children who received the highest doses continued oral feeds until the study ended at 27 and 20 months.
Seizures also appeared later and were less severe. Families reported that episodes were easier to control with standard anti-convulsant medication — a shift that translated into fewer emergency hospital visits and improved quality of life.
“These outcomes may sound modest, but for families, they are extraordinary,” said Dr. Gray-Edwards. “Eating by mouth, spending less time in hospitals, experiencing fewer seizures — these are life-changing differences.”
A Field Built on Years of Research
The trial represents the culmination of years of collaborative research by Dr. Gray-Edwards and Miguel Sena-Esteves, Ph.D., associate professor of neurology at UMass Chan. Together, they have spent much of their careers studying the biology of GM2 gangliosidosis and building gene therapy tools to counter it.
“This research reflects what our Translational Institute for Molecular Therapeutics was built to do,” said Terence R. Flotte, MD, dean of the T.H. Chan School of Medicine and senior author of the study. “We want to move discoveries from basic research into therapies that can reach patients. For families facing rare diseases, this progress represents hope where previously there was none.”
The Road Ahead
Despite the promising results, the therapy is not yet a cure. Enzyme levels did not reach the threshold needed to fully halt disease progression, and the children still faced severe neurological decline. Researchers are already working on refinements, including a single-vector delivery system that could double the amount of therapeutic DNA delivered without increasing injection volume.
The ultimate goal is to administer therapy early — ideally before symptoms begin — so that neurons can be protected before irreversible damage occurs. That may one day mean treating newborns identified through genetic screening, giving them a chance at lives free from the shadows of Tay-Sachs and Sandhoff disease.
A Glimmer of Hope
For now, the UMass Chan trial has given families something precious: proof that progress is possible. The sight of a child enjoying food months longer than expected, the sound of fewer seizures disrupting the night, the comfort of knowing that science is finally pushing back — these are small but profound victories.
“These are positive steps forward,” said Dr. Gray-Edwards. “We’re committed to continuing this work until we can provide a truly transformational therapy for these children.”
For the families living through GM2 gangliosidosis, and for generations to come, that commitment offers the most powerful medicine of all: hope.
More information: Florian Eichler et al, Dual-vector rAAVrh8 gene therapy for GM2 gangliosidosis: a phase 1/2 trial, Nature Medicine (2025). DOI: 10.1038/s41591-025-03822-4. doi.org/10.1038/s41591-025-03822-4















