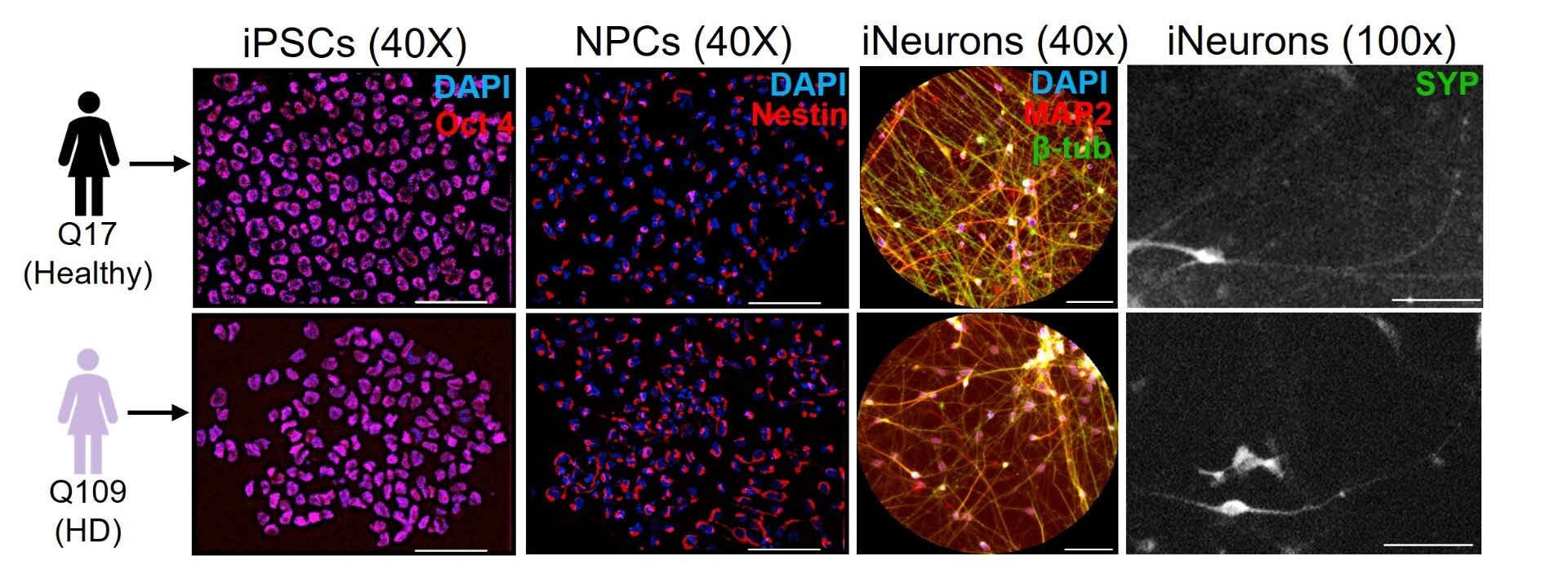
Ten years ago, researchers at the University at Buffalo took a decisive step toward understanding a devastating neurological condition that had long eluded precise explanation: Huntington’s disease. At the heart of this condition is a mutant version of a protein called huntingtin (HTT), which—like a master conductor in the brain—normally helps regulate the movement of vital cellular cargo within neurons.
What they found back then was as elegant as it was disturbing: healthy HTT functions like a traffic controller, directing vesicles and other essential materials along the neuron’s axonal highways with the help of molecular motors. But when this protein is mutated, as it is in Huntington’s disease, the traffic jams begin. Materials essential to neuronal survival become stranded. Highways are blocked. The intricate communication network that keeps the brain functioning smoothly begins to fall apart.
Now, with a new study published in Cell Death & Disease, these scientists have zoomed in on an even finer control mechanism. What if the fate of these intracellular highways isn’t just in the hands of HTT, but in the regulators that tell HTT how to behave? Enter the signal-issuing kinases: GSK3β and ERK1. These molecular managers might just be the switches we can flip to slow down or even stop the death spiral triggered by mutant HTT.
Kinases and Chaos: Regulators of Neuronal Transport
The new study, led by Dr. Shermali Gunawardena and her former Ph.D. student Thomas Krzystek, investigates two kinases—GSK3β (glycogen synthase kinase-3 beta) and ERK1 (extracellular signal-regulated kinase 1)—that seem to play opposing roles in the progression of Huntington’s disease.
Kinases are enzymes that modify other proteins by tagging them with phosphate groups in a process known as phosphorylation. These modifications act as molecular switches, turning protein functions on or off, ramping them up or down. In the case of HTT, the phosphorylation state can determine how well it performs its cargo-hauling duties along axonal tracks.
Krzystek puts it succinctly: “Kinases modify HTT and other transport components by attaching molecular tags to them known as phosphate groups.” These tags change how HTT interacts with motor proteins like kinesins and dyneins, which are the tiny, tireless engines pulling cellular cargo along microtubules.
Through a combination of cellular studies and fruit fly models, the team explored what happens when these two kinases are inhibited. The results revealed a fascinating biological paradox.
GSK3β: A Double-Edged Sword
The first kinase, GSK3β, showed itself to be a molecular saboteur when upregulated in Huntington’s-affected neurons. Inhibiting GSK3β in mutant HTT-expressing fruit fly neurons resulted in fewer axonal blockages, reduced neuronal death, and surprisingly, more agile larvae.
It was not the first time GSK3β had raised eyebrows. In earlier work, Gunawardena’s lab showed that GSK3β can dictate whether motor proteins stop or go—essentially serving as a traffic signal for the neuron’s busy roads. When GSK3β levels go awry, it’s as if all the lights turn red at once—or worse, start flashing contradictory signals. Vesicles stall. Neurons suffer. Communication fails.
“While GSK3β typically plays a positive role in neuronal function, it seems it may actually make a bad situation worse when faced with a mutant HTT,” Gunawardena explains. That statement underscores a nuanced truth in biology: a molecule’s impact depends entirely on its context. In the normal brain, GSK3β helps neurons function. In the presence of mutant HTT, it contributes to their demise.
This makes GSK3β an enticing target for therapeutic intervention. If drugs can be designed to dial down its activity—without disrupting its other essential functions—it may be possible to slow the progression of Huntington’s disease by unclogging the axonal highways and restoring some measure of cellular order.
ERK1: The Unsuspected Ally
If GSK3β is the troublemaker, ERK1 is its cautious, underrated counterpart.
When the researchers inhibited ERK1 in their fruit fly models, the effects were devastating: axonal blockages increased, and so did neuronal death. It was a stark contrast to what happened with GSK3β. ERK1 appeared to be a guardian of neuronal function in the chaotic landscape shaped by mutant HTT.
“The level of ERK1 is clearly important for Huntington’s disease, but whether it’s actually modulating the mutant HTT is unclear,” Krzystek admits. Even so, the team’s experiments left little doubt that ERK1 signaling had a protective effect in this context.
Encouraged by these findings, the researchers went a step further. What would happen if they increased ERK1 levels in the diseased fruit fly models? The results were clear: fewer blockages, fewer dying neurons, and more functional larvae. ERK1, it seemed, could serve as a neurological lifeline.
This insight, like the one about GSK3β, opened up tantalizing therapeutic possibilities. A future treatment strategy might aim to simultaneously suppress GSK3β while boosting ERK1, creating a molecular environment where HTT can function better, or at the very least, do less harm.
The Genetic Error That Starts It All
At the genetic core of Huntington’s disease lies a stretch of DNA where a sequence—cytosine-adenine-guanine (CAG)—is repeated more times than it should be in the HTT gene. These excessive repeats produce an abnormally long chain of the amino acid glutamine in the huntingtin protein, leading to toxic changes in its structure and behavior.
Normally, HTT is involved in several critical cellular processes: intracellular transport, transcription regulation, and even autophagy (the cell’s recycling system). It’s a versatile multitasker. But the mutant version tends to misfold and form aggregates, which can clog the inside of neurons, interfere with protein trafficking, and activate cell-death pathways.
Despite decades of research, scientists still don’t fully understand what normal HTT does in all its complexity—let alone why the mutated form leads to such widespread neurological breakdown. What’s becoming increasingly clear, however, is that mutant HTT disrupts early cellular events long before neurons begin to die. Catching and correcting these early changes may hold the key to slowing or even halting the disease.
From Fruit Flies to Human Hope
Why fruit flies? It might seem odd that the humble Drosophila melanogaster—a tiny insect with a brain the size of a grain of sand—would help scientists understand a human neurodegenerative disease. But the genetic tools available in flies are powerful, and many of their cellular pathways are remarkably similar to those in humans.
By manipulating kinase activity in fruit fly larvae engineered to express mutant human HTT, the researchers could observe changes in real time—both at the cellular and behavioral levels. The larvae’s ability to crawl served as a visible readout of neuronal health.
These experiments provided a rare window into the sequence of molecular events that lead from gene mutation to cell death—events that unfold invisibly inside the brains of people with Huntington’s disease over decades. Importantly, they offered something else, too: a testbed for future treatments.
Charting a Therapeutic Path Forward
“We’re trying to understand the earliest steps that ultimately lead to neurodegeneration,” Gunawardena says. “Because once a neuron dies, there’s no bringing it back. That’s why we need to intervene early—before the damage becomes irreversible.”
The concept of early intervention is especially critical in Huntington’s disease, where symptoms often don’t appear until middle age, even though mutant HTT is present from birth. Subtle cellular dysfunction may be occurring for years—perhaps even decades—before any noticeable cognitive or motor deficits emerge.
That’s why identifying molecular regulators like GSK3β and ERK1 is so valuable. They offer potential targets for drug development at a time when the disease process may still be reversible. The goal is not just to treat symptoms, but to address root causes and preserve neuron function for as long as possible.
Beyond Huntington’s
Though the current study focuses specifically on Huntington’s disease, the implications may stretch much further. Dysregulated axonal transport has been implicated in other neurodegenerative conditions, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS). The idea that fine-tuning kinase activity could correct transport defects may one day be applicable across a spectrum of disorders.
Moreover, the idea that cellular dysfunction precedes neuronal death in these diseases shifts the paradigm in how we think about treatment. We’re not just chasing symptoms anymore—we’re hunting for the molecular saboteurs that start the breakdown.
A Glimpse of Control Amid the Chaos
The human brain is a marvel of organized chaos. Billions of neurons fire, signal, and transmit messages across dizzyingly complex networks. That this whole system can be thrown into disarray by a few repeated bits of genetic code is a sobering reminder of our biological fragility.
But in that fragility lies a kind of beauty: the possibility that a molecule here or a protein there can make all the difference. That silencing a single disruptive signal—or boosting a protective one—can restore balance.
As Dr. Gunawardena’s team continues to unravel the tangled webs of neuronal transport, their work offers not just a deeper understanding of Huntington’s disease, but a glimpse of hope. Perhaps one day, by controlling the cellular traffic, we can save the passengers—the neurons—and with them, the lives and memories of those at risk.
Reference: Thomas J. Krzystek et al, Opposing roles for GSK3β and ERK1-dependent phosphorylation of huntingtin during neuronal dysfunction and cell death in Huntington’s disease, Cell Death & Disease (2025). DOI: 10.1038/s41419-025-07524-0