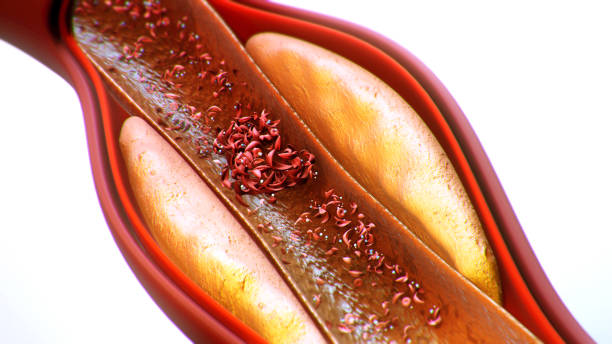
Imagine the body as a vibrant city, with blood as its bustling traffic network carrying oxygen—the fuel of life—to every street and corner. For most people, the red blood cells glide smoothly like cars on a well-paved highway, round and flexible, able to squeeze through even the narrowest streets. But for those living with sickle cell disease, these cells take on the shape of a crescent moon, stiff and sticky, more like a broken-down vehicle clogging the road.
This change may seem small, but its consequences are profound. Roads get blocked, oxygen delivery stalls, and the whole city begins to suffer. The pain is not only physical but also emotional and social, as those living with the disease face lifelong challenges. Yet, alongside the hardships is a story of resilience, of groundbreaking science, and of communities coming together to fight for better treatments and brighter futures.
To understand sickle cell disease fully, we must travel deep into its genetic roots, explore the symptoms it brings, and examine how science is working tirelessly to ease its burden.
What is Sickle Cell Disease?
Sickle cell disease (SCD) is a group of inherited blood disorders that primarily affect hemoglobin, the protein inside red blood cells that carries oxygen throughout the body. Instead of producing normal, round red blood cells, people with SCD produce cells shaped like sickles or crescent moons. These cells are rigid, sticky, and fragile.
Because of their shape, sickle cells:
- Break apart easily, leading to anemia (a shortage of healthy red blood cells).
- Get stuck in small blood vessels, blocking the flow of oxygen.
- Cause sudden episodes of pain, organ damage, and other serious health problems.
SCD is not a single condition but rather a collection of related disorders. The most common and severe type is sickle cell anemia (HbSS), where a person inherits two sickle genes—one from each parent. Other forms include HbSC disease and sickle beta-thalassemia, which result from different genetic combinations.
The Genetic Cause: A Tiny Mutation with Massive Impact
At the heart of sickle cell disease is a single genetic mutation in the HBB gene on chromosome 11. This gene provides instructions for making the beta-globin subunit of hemoglobin. Normally, hemoglobin is composed of four protein chains—two alpha and two beta chains—that work together to carry oxygen.
In SCD, the mutation changes just one building block (amino acid) in the beta-globin chain: valine replaces glutamic acid. This tiny change leads to the production of hemoglobin S instead of normal hemoglobin A.
When hemoglobin S releases oxygen, it tends to stick together, forming long, rigid strands inside the red blood cell. This causes the cell to twist into the characteristic sickle shape. Unlike normal red cells that live for about 120 days, sickle cells break apart after just 10–20 days, leading to chronic anemia.
This mutation arose thousands of years ago in regions where malaria was common, such as Africa, the Middle East, and parts of India. Carriers of just one sickle cell gene (known as the sickle cell trait) are resistant to severe malaria. This evolutionary advantage explains why the gene persists, even though inheriting two copies results in disease.
Symptoms: The Human Experience of Sickle Cell Disease
The symptoms of sickle cell disease vary widely among individuals. Some may experience mild discomfort, while others face life-threatening complications. The unpredictability of the disease is one of its most challenging aspects, but several hallmark features define it.
Chronic Anemia
Because sickle cells die so quickly, the body cannot replace them fast enough. This leads to anemia, which manifests as fatigue, weakness, pale skin, and delayed growth in children.
Pain Crises
Perhaps the most well-known symptom of SCD is the pain crisis, also called vaso-occlusive crisis. When sickle cells clump together and block blood vessels, oxygen cannot reach tissues. The result is sudden, intense pain that may last hours to days. These crises often require hospitalization and are a major cause of suffering.
Organ Damage
Over time, repeated blockages and chronic anemia damage organs:
- The spleen, which filters blood, often becomes scarred and shrinks, leaving patients vulnerable to infections.
- The kidneys may fail to concentrate urine properly, leading to dehydration and eventual kidney disease.
- The liver, heart, lungs, and brain can also be damaged.
Infections
Because the spleen is compromised, people with SCD are more prone to bacterial infections such as pneumonia, meningitis, and sepsis. Infections remain a leading cause of death in children with the disease.
Stroke
Blocked blood flow in the brain can trigger strokes, especially in children and young adults. Without preventive care, this complication affects up to 10% of children with SCD.
Delayed Growth and Puberty
The shortage of oxygen and nutrients slows normal growth, causing children to be shorter and enter puberty later than their peers.
Vision Problems
Tiny blood vessels in the eyes can become blocked, leading to retinal damage and potential blindness.
The combination of these symptoms creates a disease that affects nearly every system of the body, often in unpredictable ways.
Diagnosis: How Sickle Cell Disease is Detected
Early and accurate diagnosis is critical for managing SCD. Many countries now screen newborns for the condition so treatment can begin immediately.
Newborn Screening
A simple blood test can identify abnormal hemoglobin. In the United States and many other nations, this is standard practice. Infants diagnosed early can receive preventive care to reduce infections and other complications.
Blood Tests
Other tests help confirm the diagnosis:
- Hemoglobin electrophoresis separates different types of hemoglobin, showing the presence of hemoglobin S.
- Genetic testing identifies mutations in the HBB gene.
- Complete blood count (CBC) reveals anemia and other blood abnormalities.
Imaging and Monitoring
Once diagnosed, patients may undergo additional tests to monitor complications, such as transcranial Doppler ultrasound to assess stroke risk, or echocardiograms to evaluate heart function.
Treatment: From Symptom Relief to Curative Approaches
Although sickle cell disease is lifelong, advances in medicine have greatly improved outcomes. Treatment strategies range from managing symptoms to potentially curing the disease.
Pain Management
Pain crises are treated with hydration, oxygen, rest, and medications. Mild pain may be relieved with over-the-counter drugs, but severe crises often require opioids under medical supervision.
Preventing Infections
Children with SCD are often given daily penicillin until at least age 5 to protect against bacterial infections. Vaccinations—especially against pneumococcus, meningococcus, and influenza—are essential.
Blood Transfusions
Regular blood transfusions reduce the proportion of sickle cells in circulation, lowering the risk of stroke and other complications. However, long-term transfusions carry risks, including iron overload, which must be managed with chelation therapy.
Hydroxyurea
This medication stimulates the production of fetal hemoglobin (HbF), which does not sickle. By increasing HbF levels, hydroxyurea reduces pain crises, acute chest syndrome, and the need for transfusions. It is now widely used in both children and adults with SCD.
Bone Marrow and Stem Cell Transplants
Currently, the only established cure for SCD is a bone marrow or stem cell transplant from a compatible donor, often a sibling. The transplanted stem cells can produce normal red blood cells, effectively eliminating the disease. However, transplants carry risks, including rejection and severe infections, and are not available to all patients.
Emerging Therapies: Gene Editing and Beyond
The future of SCD treatment is filled with hope. Cutting-edge techniques like CRISPR-Cas9 gene editing are being tested to correct the defective gene or boost the production of fetal hemoglobin. Clinical trials show promising results, raising the possibility that gene therapy may one day provide a safe, widely available cure.
Living with Sickle Cell Disease: The Human Side
Beyond medical treatments, living with SCD requires daily resilience. The disease can limit school attendance, work opportunities, and social activities. Many patients face stigma, particularly when seeking pain relief in hospitals where opioid use can be misunderstood.
Support from family, communities, and advocacy organizations is vital. Counseling and mental health care help individuals cope with chronic pain and uncertainty. Education about the disease—both for patients and the public—reduces stigma and improves outcomes.
Importantly, many people with SCD are leading full, meaningful lives. With proper care, life expectancy has increased dramatically, and patients are pursuing careers, raising families, and advocating for change.
Global Burden of Sickle Cell Disease
SCD affects millions worldwide, particularly in sub-Saharan Africa, India, the Middle East, and among people of African descent in the Americas and Europe. In Africa alone, over 300,000 babies are born with the condition each year, and many do not survive childhood due to lack of healthcare resources.
The global burden highlights stark inequalities in healthcare access. While children in wealthier countries often live into adulthood, those in poorer regions face high mortality rates. Expanding newborn screening, access to antibiotics, vaccines, and affordable medications is critical for saving lives.
Hope for the Future
The story of sickle cell disease is not only about pain and struggle but also about resilience and hope. From a single genetic mutation that arose as protection against malaria has come both suffering and scientific discovery.
Today, thanks to research and advocacy, the outlook is brighter than ever. Life expectancy is increasing, new therapies are on the horizon, and the voices of patients are being heard. What once seemed like an insurmountable burden is now being met with innovation and compassion.
The road ahead will not be easy, especially in addressing global health disparities. But every step forward—every child vaccinated, every new therapy tested, every life saved—is part of a larger movement toward justice and healing.
Conclusion: Defining Sickle Cell Disease Beyond Genetics
Sickle cell disease is more than a genetic disorder. It is a human story, shaped by biology, culture, and resilience. It begins with a microscopic change in hemoglobin but expands outward, touching every organ, every family, and entire communities.
Understanding its causes reveals the incredible power of a single gene. Recognizing its symptoms shows us the complexity of human suffering. Exploring its treatments highlights the creativity of science and medicine. And listening to the voices of those living with the disease reminds us that health is not only a medical condition but also a lived experience.
As science moves closer to cures and societies push for equality in healthcare, the story of sickle cell disease is evolving. It is a story of pain, yes—but also of hope, courage, and the possibility of transformation.





