For patients living with sickle cell disease, life is often marked by unpredictable pain, exhaustion, and the slow, invisible wear of a body constantly at war with itself. But among the many complications that haunt this inherited condition, one stands out for its speed and severity: acute chest syndrome (ACS). It’s a terrifying, often fatal crisis where every breath feels like inhaling through broken glass. Despite being the leading cause of death in sickle cell patients, ACS remains a medical enigma—one for which doctors have few tools beyond oxygen masks, painkillers, and hope.
But now, in a study that may rewrite how we understand and treat this life-threatening condition, researchers at Mass General Brigham have discovered a surprising culprit: an ancient immune system pathway called the complement system. The study, published in Science Translational Medicine, offers the first concrete evidence that this overlooked arm of the immune response plays a central role in both triggering and amplifying ACS. And, perhaps even more importantly, it offers a glimpse of hope in the form of existing drugs that could interrupt this deadly chain reaction.
“We’ve known for a long time that ACS is dangerous, but we haven’t truly understood why it happens or how to stop it,” says Dr. Sean Stowell, senior author of the study and a physician-scientist in the Department of Pathology at Brigham and Women’s Hospital. “Now, we’re starting to see the biological dominoes falling into place.”
The Silent Saboteur Within
Sickle cell disease begins with a single letter change in a gene that shapes hemoglobin—the protein that carries oxygen in red blood cells. That small genetic typo has devastating consequences. It warps red blood cells into rigid, sickle-like shapes that are prone to breaking apart and blocking blood vessels. This destruction, known as hemolysis, triggers waves of inflammation, pain, and organ damage across the body.
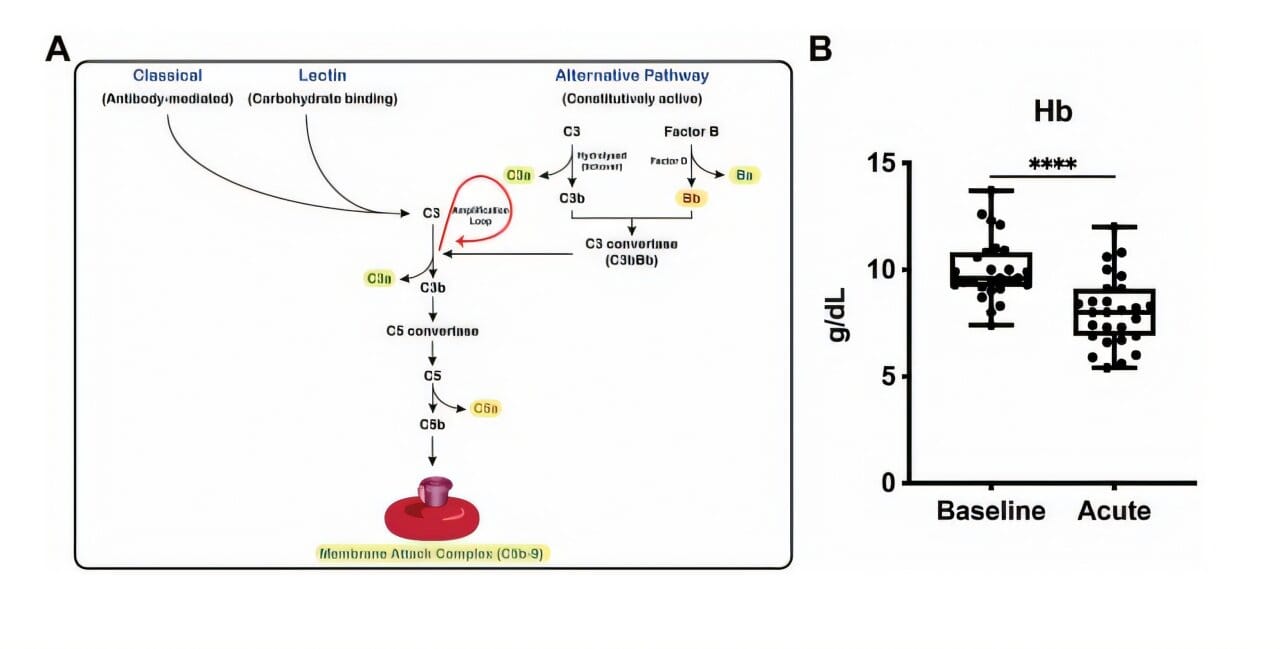
For decades, hemolysis was viewed primarily as a consequence of the disease—just a symptom. But this new research shows that it may also be part of a dangerous feedback loop. When red blood cells break down, they release materials that activate the complement system: a set of proteins that make up one of our body’s oldest immune defense tools, predating even antibodies.
Normally, the complement system helps destroy bacteria and viruses. But in the context of sickle cell disease, it appears to be misfiring—attacking the body’s own red blood cells, amplifying their destruction, and pouring fuel on an already raging fire. This cycle doesn’t just intensify symptoms. It helps create ACS.
“Our results suggest that the complement system, once activated, doesn’t stop at simply responding to damage,” says Dr. Stowell. “It becomes a key player in worsening the very disease it’s trying to fight.”
Clues in the Blood
To trace the outlines of this hidden mechanism, the researchers began with a simple question: do sickle cell patients show signs of abnormal complement activity, especially during episodes of ACS?
Blood samples from 27 individuals with sickle cell disease provided a sobering answer. Even at baseline—meaning, when patients weren’t in the middle of a health crisis—their blood contained significantly higher levels of activated complement proteins compared to healthy individuals. But during ACS, the complement system went into overdrive, producing even greater quantities of these proteins.
That alone was a red flag. But what made the findings even more compelling was what happened next. Using genetically engineered mice that mimic the key features of sickle cell disease, the scientists saw a clear pattern: when complement activation was high, so was red blood cell destruction. When they disrupted this pathway—either by removing specific complement proteins or by using drugs that block them—the destructive cycle slowed, and symptoms of ACS diminished.
“It’s like discovering a hidden hand tipping the balance toward crisis,” says Dr. Yasmine Belhassen, a co-author of the study. “The complement system is supposed to protect us, but here, it becomes part of the pathology.”
A Feedback Loop With Fatal Consequences
What emerged from the study was a disturbing chain reaction. Sickle-shaped red blood cells are already fragile. When they break apart—through stress, low oxygen, or random chance—they release hemoglobin and other molecules that aren’t meant to be loose in the bloodstream. These molecules activate the complement system.
Once activated, the complement system doesn’t just clean up the mess. It attacks nearby red blood cells, even those that haven’t yet sickled, increasing hemolysis. This leads to more inflammation, more immune activation, and more cellular destruction—creating a deadly loop that ends in ACS, where the lungs become inflamed, oxygen levels plummet, and life hangs in the balance.
In this context, ACS is not just a respiratory emergency. It is the tip of a biochemical iceberg—one that begins with a genetic mutation but crashes into ancient immune systems gone awry.
The Hope Inside the Hypothesis
If the complement system is helping drive ACS, could it also be stopped?
Here, the study offers cautious but thrilling optimism. There are already drugs on the market that target various components of the complement system. These include medications approved for rare conditions like paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS)—both diseases involving complement-driven destruction of red blood cells.
In preclinical animal models, researchers used these same types of drugs to block key complement proteins. The results were dramatic. Not only did the drugs reduce hemolysis, they also protected against the development of ACS-like symptoms. The mice breathed more easily. Their lungs showed fewer signs of damage. The cascade had been interrupted.
“The fact that these drugs already exist—and are approved for other conditions—gives us a running start,” explains Dr. Stowell. “It means we’re not starting from scratch. We can begin designing clinical trials much sooner.”
Of course, this doesn’t mean a cure is around the corner. Mice are not people, and human biology is vastly more complex. Still, the path forward is clearer now than ever before. The researchers believe that randomized clinical trials using complement inhibitors—each targeting different parts of the pathway—could help determine which therapies are most effective, and when to deploy them.
A Shift in Perspective
Perhaps the most profound implication of the study is conceptual, not therapeutic. For years, acute chest syndrome was viewed as an inevitable complication of sickle cell disease—something that happened when too many red blood cells clogged the lungs or when infection struck at the wrong time.
But this research tells a different story. It suggests that ACS is not just the product of passive breakdown, but of an active immune process that can be influenced, interrupted, and potentially even prevented.
It recasts ACS not as a sudden thunderstorm but as a predictable storm system, one that can be mapped and, eventually, diverted.
The Human Side of the Crisis
Behind every data point in this study is a human being—a child gasping for breath, a parent pacing in the hospital corridor, a teenager whose lungs feel like they’re filling with fire. For patients with sickle cell disease, ACS is not an abstraction. It is a moment when everything hangs in the balance.
That’s why these findings matter. Because while science often marches forward in cautious steps, for families facing this disease, every advance brings a ripple of hope. Hope that one day, the ER won’t be the only answer. That instead of waiting for lungs to collapse, doctors can intervene early, armed with knowledge and medicine tailored to the body’s ancient signals.
“This is personal,” says Dr. Stowell. “We’re not just studying a disease. We’re trying to end one of its most devastating outcomes.”
Looking to the Horizon
As this work advances into the next stage—clinical trials, human testing, real-world evaluation—the scientific community will be watching closely. But so will patients. So will parents. So will entire communities that have long borne the weight of this disease with too little recognition and too few breakthroughs.
This discovery doesn’t erase that history. But it writes a new chapter—one where understanding replaces mystery, and possibility begins to rise where there was once only pain.
And in that chapter, perhaps, ACS will no longer be a dreaded inevitability—but a crisis we can predict, prevent, and, one day, leave behind.
Reference: Satheesh Chonat et al, Complement is activated in patients with acute chest syndrome caused by sickle cell disease and represents a therapeutic target, Science Translational Medicine (2025). DOI: 10.1126/scitranslmed.adl4922