Every year, millions of patients put their faith in the cutting-edge advances of modern oncology. Chemotherapy, radiation, targeted therapy—these weapons are forged in the crucible of science, designed to destroy the enemy that grows within: cancer. Yet, far too often, the story doesn’t end with remission. The disease returns—stronger, smarter, resistant.
This cruel twist isn’t just a medical problem. It’s a human one. Cancer resistance is what turns treatable diagnoses into fatal ones. It’s the shadow that looms behind every successful treatment. Scientists estimate that treatment resistance is responsible for up to 90% of cancer-related deaths, a staggering statistic that reveals a cold, unyielding truth: cancer doesn’t just mutate—it adapts.
But what if we could decode the secret? What if we could find out exactly how cancer cells continue to multiply even after their DNA has been damaged—supposedly beyond repair—by treatments designed to stop them in their tracks?
A research team from the University of Ottawa Faculty of Medicine, led by Dr. Damien D’Amours and graduate student Laurence Langlois-Lemay, has just taken a major step toward that goal. Their recent discovery, published in the prestigious journal Proceedings of the National Academy of Sciences (PNAS), uncovers a surprising mechanism by which damaged cells escape destruction and continue dividing. It may be one of the most important keys yet to understanding—and ultimately defeating—cancer resistance.
When DNA Damage Should Spell the End
The backbone of many cancer therapies lies in damaging the DNA of tumor cells. It’s a high-stakes gamble: induce so much genetic harm that the cells either self-destruct or become too dysfunctional to reproduce. And for a time, this works. Tumors shrink. Symptoms recede. Hope rises.
But cancer, unlike most diseases, doesn’t play by the rules. Sometimes, the very DNA damage intended to destroy it becomes a trigger for the disease’s rebirth. The new study out of Ottawa explains how.
In healthy cells, there are built-in security checkpoints—places in the cell cycle where the system pauses to evaluate whether it’s safe to proceed with division. If DNA damage is detected, these checkpoints activate repair mechanisms, or if the damage is too extensive, they send the cell into apoptosis, a kind of programmed suicide. It’s a beautifully designed process, a molecular ethics system designed to keep chaos at bay.
But what happens when the cell refuses to listen? What if a damaged cell finds a way to override the warning signs and barrels forward anyway, copying its corrupted code and passing it on to its descendants?
That’s precisely what Dr. D’Amours’ team has found—an elegant, troubling answer to why treatment-resistant tumors continue to grow.
A Surprising Culprit: The Centrosome
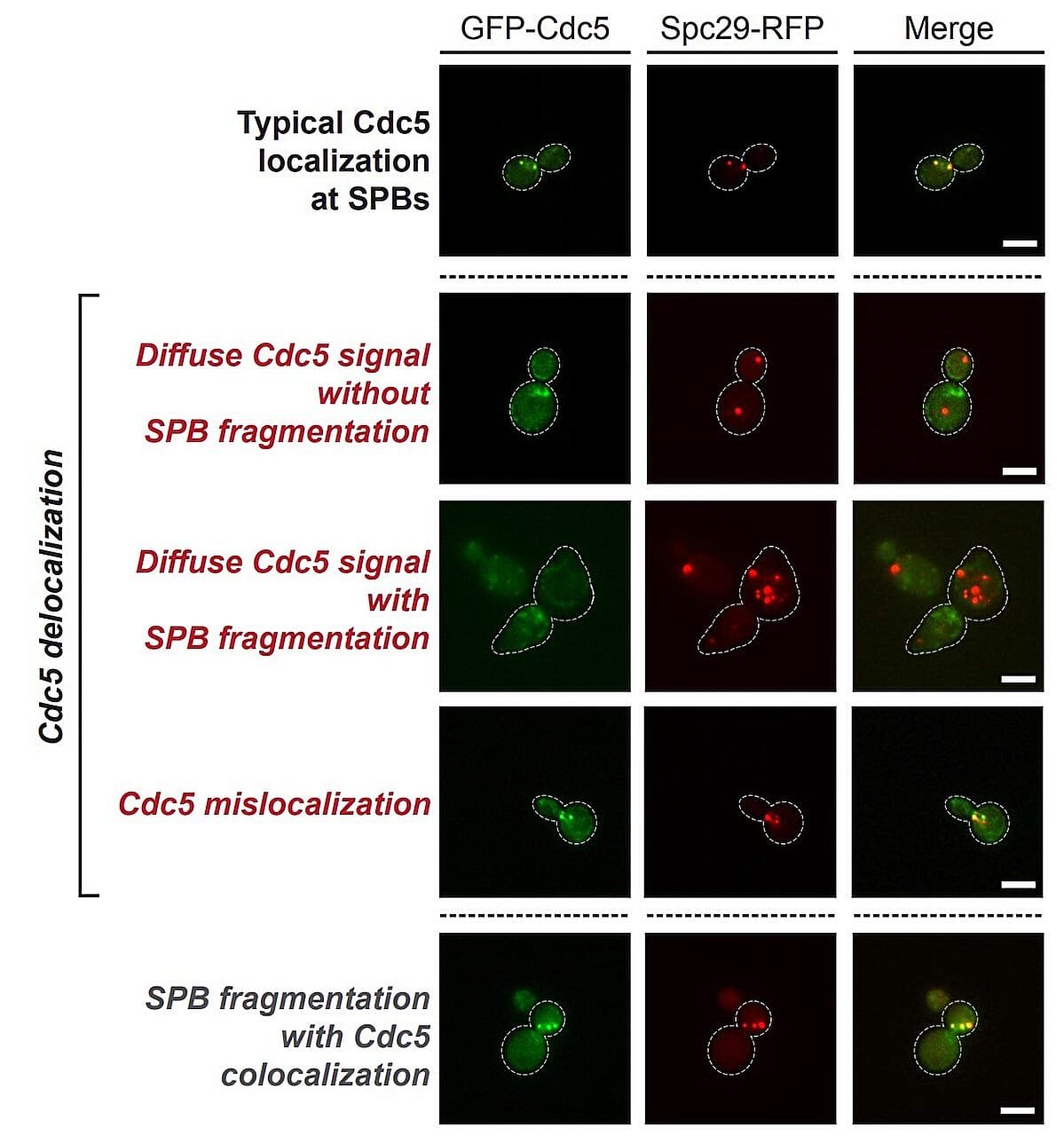
At the heart of this discovery is a tiny, overlooked structure inside cells called the centrosome. Historically, centrosomes have been seen as the cell’s organizational anchors during division, pulling chromosomes apart like a biological tug-of-war. But the research from Ottawa reveals they’re far more cunning than previously imagined.
Dr. D’Amours’ lab has shown that centrosomes serve as critical decision-making hubs, particularly in damaged cells. When DNA is irreparably injured—say, by chemotherapy or radiation—cells are supposed to stop. But the centrosomes, guided by specific molecular signals, can choose to override this.
A major player in this override mechanism is an enzyme called Polo-like kinase 1 (PLK1), a known regulator of cell cycle progression. The researchers discovered that when PLK1 is recruited in large quantities to the centrosomes, it essentially tells the cell, “Ignore the damage. Keep dividing.” And so it does—despite the dangerous mutations it carries. This is how resistance begins.
“Cell proliferation in the presence of DNA lesions is arguably the most effective way to generate cancers in humans,” Dr. D’Amours explains. “Ramped-up recruitment of PLK1 at centrosomes promotes adaptation to DNA damage, leading to aberrant cell proliferation and its pro-oncogenic effects.”
In simpler terms: the cell overrides its own emergency brakes, and cancer drives right through them.
The Yeast That Told a Human Story
The road to this discovery began not with cancer patients, but with yeast—a tiny organism that has, for decades, served as a genetic model for understanding complex cellular processes in humans. Why yeast? Because it shares many of the same genetic and cellular functions as we do, including how it handles DNA damage.
Langlois-Lemay, then a master’s student, started by modeling the DNA damage adaptation response in yeast using a systems biology approach—a kind of holistic view that lets researchers see how molecular systems interact as a whole, rather than in isolated parts.
Her early work revealed a critical component within the yeast centrosome that allowed PLK to gather and launch the cell into division—even when its DNA was severely damaged. This component, they later found, is evolutionarily conserved—meaning it has remained unchanged through hundreds of millions of years, from yeast to humans.
This astonishing continuity across species offered compelling evidence that a similar process could be happening in human cancer cells. And it raised a thrilling—and terrifying—possibility: if cancer hijacks this ancient cellular mechanism, could we hijack it back?
A Molecular Lever Against Resistance
Understanding that centrosomes and PLK1 play this crucial role in enabling cancer cells to survive treatment opens a new path for medicine. By chemically inhibiting PLK1, it may be possible to restore the cell’s natural sensitivity to DNA damage—forcing damaged cancer cells to either stop dividing or die, as they should.
Dr. D’Amours emphasizes the potential of this strategy as a complement to traditional therapies. “Our discoveries provide crucial insights into how cancer cells resist clinical treatments based on radio/chemotherapy-induced DNA damage,” he says. “Drug-based inhibition of PLK1 represents a promising strategy to prevent adaptation to therapy-induced DNA damage.”
The implications of this are vast. If validated in clinical settings, PLK1 inhibitors could be used in tandem with standard cancer therapies, acting like a molecular gatekeeper that ensures the treatment does what it’s supposed to—halt cancer in its tracks.
The Long Road to a Breakthrough
None of this came easily. The work began in 2017, when Dr. D’Amours moved his lab from Université de Montréal to the University of Ottawa. There, with the support of the Ottawa Institute of Systems Biology, he and his team built the infrastructure to ask some of the deepest, most fundamental questions about how cells make decisions in the face of genetic catastrophe.
For Langlois-Lemay, now completing her Ph.D., the journey has been intensely personal. “She chipped away at this project for years,” says Dr. D’Amours. “What she’s accomplished is a real success story—not just for her, but for the entire field.”
This kind of patient, methodical research—years of microscopic observation, modeling, experimentation—doesn’t often make headlines. But it’s the kind of work that builds the foundation for real medical revolutions.
From the Lab Bench to the Hospital Bed
The next step for the Ottawa team is to move beyond yeast and cell lines into clinical research, examining how these mechanisms operate in human tissues and tumors. If PLK1’s role in treatment resistance holds true in patients, it could pave the way for new combination therapies, customized to block the very escape routes cancer uses to survive.
This is what’s known as bench-to-bedside translation—the gold standard of biomedical research. It means taking what was discovered under a microscope and using it to extend someone’s life, relieve their suffering, or cure their disease.
And for the millions who face cancer every year, this kind of translation could mean the difference between relapse and remission.
Hope Written in the Code
In the end, cancer isn’t just a disease of cells. It’s a disease of miscommunication—a conversation gone awry at the molecular level, where signals meant to protect the body are misread, ignored, or deliberately overridden.
This new research helps decode one of the most dangerous of those miscommunications. It offers not just an explanation, but a way forward. A strategy. A lever we can pull.
And for all its microscopic detail and biochemical nuance, the story at its heart is profoundly human: it’s the story of how we try to outthink the enemy within, not just with power, but with understanding. Because in science, as in life, real victory rarely comes from force alone. It comes from learning how the world works—cell by cell, signal by signal—and using that knowledge to heal.
Reference: Laurence Langlois-Lemay et al, Yeast centrosomes act as organizing centers to promote Polo kinase–mediated adaptation to persistent DNA damage, Proceedings of the National Academy of Sciences (2025). DOI: 10.1073/pnas.2414426122