In the world of pharmaceuticals, a stealthy sabotage often unfolds inside our cells. The culprits? A family of proteins known as cytochrome P450 (CYP) enzymes, which are responsible for breaking down a staggering 80% of all FDA-approved drugs. Their role is essential — they help metabolize foreign substances to protect the body. But there’s a catch: in doing so, they also dismantle life-saving medications, weakening their potency right when patients need them most.
Now, in a groundbreaking leap forward, researchers at St. Jude Children’s Research Hospital have cracked one of pharmacology’s most frustrating puzzles: how to stop CYP3A4, a particularly voracious member of the CYP family, from chewing up critical drugs — without accidentally hobbling other important enzymes in the process. The solution? A set of newly designed drug frameworks that zero in on CYP3A4 with surgical precision, potentially rewriting how we design and deliver medications in the future.
The full findings, published in Nature Communications, don’t just unveil new inhibitors — they map out the very structural blueprint for overcoming off-target effects that have plagued past attempts.
Why CYP3A4 Is Both Hero and Villain
CYP3A4 is the molecular multitasker of the liver, tasked with detoxifying foreign compounds — including many of our most vital drugs. It metabolizes cancer treatments like paclitaxel, antivirals such as nirmatrelvir (a key component in Paxlovid), and countless others.
Doctors often co-administer CYP3A4 inhibitors to boost the effectiveness of drugs by slowing down their breakdown. One famous example is ritonavir, which is paired with nirmatrelvir to keep its levels high in the body when fighting COVID-19.
But here’s the problem: current CYP3A4 inhibitors are blunt instruments. They don’t just inhibit CYP3A4 — they often hit other CYPs, including CYP3A5, a close relative with similar structure and function. This collateral damage can be catastrophic, leading to toxic drug accumulation, dangerous side effects, or even life-threatening interactions.
“Unintended inhibition of CYPs can drastically affect how drugs behave in the body,” explains Dr. Taosheng Chen, Ph.D., PMP, corresponding author of the study and a leading researcher at St. Jude’s Department of Chemical Biology & Therapeutics. “When you use a nonselective CYP3A inhibitor, you run the risk of pushing plasma drug levels into dangerous territory.”
From Thousands to Three: Precision Through High-Throughput Screening
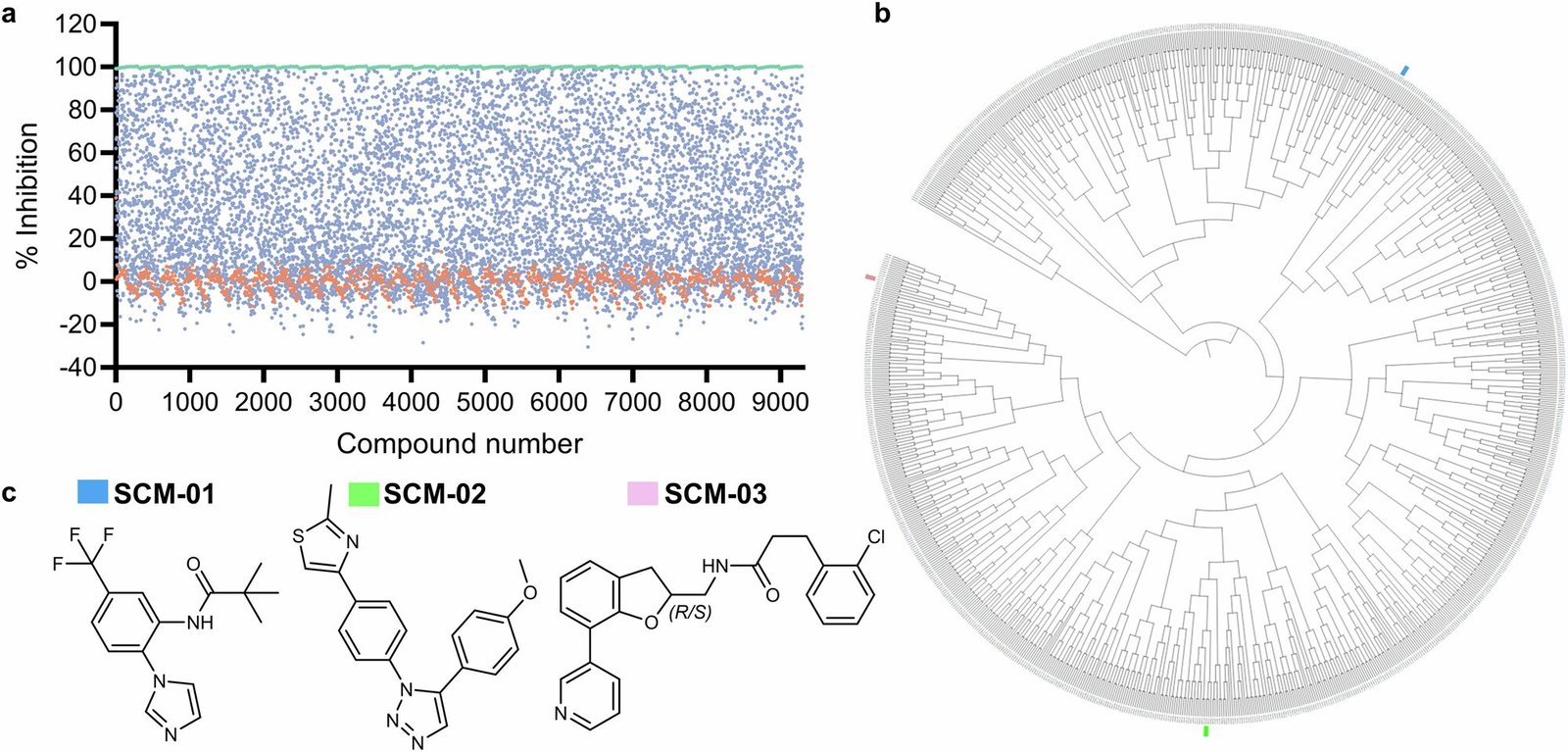
To tackle this problem, the research team embarked on an ambitious project — combing through 9,299 potential inhibitor compounds using high-throughput screening technology. Their goal? To find inhibitors that could lock onto CYP3A4 while leaving CYP3A5 — and other CYPs — untouched.
Through a rigorous filtering process, they whittled the list down to just three potent scaffolds. These chemical frameworks showed promising selectivity for CYP3A4 without binding to its biological siblings.
But how exactly did these molecules achieve such rare precision?
Peering Into the Pocket: What X-Ray Crystallography Revealed
To answer that, the team turned to X-ray crystallography, a technique that allows researchers to visualize the atomic structure of proteins in exquisite detail.
What they found was illuminating: although CYP3A4 and CYP3A5 share many structural features, there’s a critical difference — a loop at the end of the CYP3A5 protein (its C-terminus) that physically blocks the binding site.
“There is a narrower binding pocket for CYP3A5,” said Chen. “This loop acts like a molecular gate, stopping certain inhibitors from getting in. That’s why some compounds can slip into CYP3A4 but get repelled by CYP3A5.”
This difference, once just a minor structural footnote, suddenly became the cornerstone of the team’s strategy. By tweaking their inhibitors to exploit this spatial mismatch, they crafted molecules that fit snugly into CYP3A4’s open binding pocket — but couldn’t breach CYP3A5’s tighter defenses.
Meet SCM-08: A Molecular Marksman
Among the newly designed compounds, one stood out: SCM-08, a highly refined inhibitor that exhibited an astonishing 46-fold preference for CYP3A4 over CYP3A5.
Even more impressively, SCM-08 avoided interactions with other off-target CYP proteins that typically get swept up by conventional inhibitors. This kind of specificity is virtually unheard of in the field — and it opens the door to a new generation of precision-guided drugs.
“SCM-08 is more than just a promising compound,” Chen said. “It’s a launching point. We now have a structural basis for building even better inhibitors. We can increase potency while maintaining — or even enhancing — selectivity.”
What This Means for the Future of Drug Development
The implications of this discovery ripple far beyond CYP3A4. With the structural insights gleaned from this work, researchers now have a template for targeting other tricky CYP enzymes — potentially minimizing adverse drug interactions across a range of therapeutic areas.
This could be transformative for fields like:
- Oncology, where maintaining precise drug levels is critical to avoid resistance and toxicity.
- Infectious diseases, where viral therapies are often combined with enzyme inhibitors.
- Personalized medicine, where genetic variations in CYP enzymes can dramatically alter drug metabolism.
Moreover, the ability to selectively control CYP3A4 activity could improve how drugs are dosed and combined, reducing the need for higher, more toxic doses simply to counteract metabolism.
Toward a New Era of Metabolic Control
What St. Jude’s team has accomplished is nothing short of a paradigm shift. For decades, researchers have wrestled with the challenge of inhibiting drug-metabolizing enzymes without collateral damage. Now, with the unveiling of SCM-08 and its underlying architecture, we’re witnessing the first steps into an era where enzyme inhibitors are not blunt tools, but scalpel-sharp instruments.
This study is more than a win for structural biology or chemical therapeutics — it’s a triumph of rational drug design, the ability to tailor molecules with such finesse that they navigate the crowded landscape of the cell like guided missiles.
In the hands of future drug developers, these insights could become the foundation for safer, smarter, and more effective therapies. And at the heart of it all is a deceptively simple idea: understand the structure, and you can master the function.
The next time you take a drug, it may owe its strength not just to the active ingredient, but to the elegant science that ensures it stays active — right where it counts.
Reference: Jingheng Wang et al, Decoding the selective chemical modulation of CYP3A4, Nature Communications (2025). DOI: 10.1038/s41467-025-58749-8